Difference between revisions of "Förster Resonance Energy Transfer"
(→Orientation Factor and Overlap Integral) |
(→Orientation Factor and Overlap Integral) |
||
| Line 167: | Line 167: | ||
<math>f_{D}(\lambda )=\frac{F_{D\lambda }(\lambda )}{\int F_{D\lambda }d\lambda } \tag{7}</math> | <math>f_{D}(\lambda )=\frac{F_{D\lambda }(\lambda )}{\int F_{D\lambda }d\lambda } \tag{7}</math> | ||
| + | |||
| + | where <math>F_{D\lambda }</math> is the donor fluorescence per unit of wavelength interval and the integral extends over the relevant donor emission band(s). The overlap integral can alternatively be expressed in wavenumber form or in frequency form [1]. | ||
Revision as of 00:24, 16 December 2017
FRET http://photobiology.info/PDF/History%20of%20FRET.pdf https://link.springer.com/article/10.1140%2Fepjh%2Fe2013-40007-9
by Dr. B. Wieb VanDerMeer, Department of Physics and Astronomy, Western Kentucky University, Bowling Green, KY 42101-1077, USA.
FRET is Fluorescence with Resonance Energy Transfer, or Fluorescence-detected Resonance Energy Transfer, or Förster Resonance Energy Transfer. It is an absorption of electromagnetic radiation by one molecule, the “donor,” followed by resonance interaction with another molecule, the “acceptor.” As a result, energy is transferred from donor to acceptor. The two molecules can be different or identical. If the two molecules are different, the process is called “hetero-transfer” (illustrated in Figure 1). If the two are identical we speak of “homo-transfer” (illustrated in Figure 2) and the energy can go back and forth for a while or go to yet another molecule until emission or a radiation-less transition takes place. FRET is extremely sensitive to the distance between the interacting molecules and can be used to visualize interactions and to measure distances between 1 and 10 nanometers.


FRET can be observed in different ways. A convenient method in the case of hetero-transfer is to examine the effect of FRET on spectra of donors and acceptors as illustrated in Figure 3.

Contents
1 Acronym
The most frequently used interpretation of the acronym FRET is “Fluorescence Resonance Energy Transfer,” which is incorrect, because fluorescence is not transferred. Correct readings of FRET are “Fluorescence with Resonance Energy Transfer” (preferred by the present author [1]), or “Fluorescence-detected Resonance Energy Transfer” [2], or “Förster Resonance Energy Transfer “ [3, 4]. The phenomenon itself is Resonance Energy Transfer. RET or Resonance Energy Transfer used to be the most popular acronym [5]. Fluorescence serves to detect RET. Förster is the main force behind FRET.
2 Förster’s Contributions

Förster’s contributions to FRET are references 10 through 15. His 1946-paper [10] is a short preview of his 1948-paper [11]. In this preview he states the main idea in clear non-mathematical language. The 1948-paper is his most cited paper. There he focuses on FRET between like molecules (homotransfer). This type of transfer can be detected as a decrease in the polarization or anisotropy of the fluorescence with increasing concentration. Reabsorption followed by secondary emission contributes to depolarization, but it can only account for a fraction of the observed depolarization. He introduces the separation [math]R_{0}[/math] (later called the Förster distance or Förster radius; he himself called it the “critical molecular separation”), the separation distance between the donor and the acceptor at which the probability for transfer equals the combined probabilities for all other ways to deactivate the excited donor, and expresses this distance in terms of the overlap integral, the index of refraction of the medium, and the lifetime without transfer. From his data and data in the literature he calculates for fluorescein to fluorescein in water: [math]R_{0}[/math]= 5.0 nanometer, and for chlorophyll A to chlorophyll A in ethyl ether: [math]R_{0}[/math]= 8.0 nanometer. He also defines a critical concentration, [math]c_{0}[/math]c0, as the product of known constants and the inverse cube of [math]R_{0}[/math]. He made the simplification (actually, a very good approximation) that only photons emitted by the primary molecule are maximally polarized, and the ones emitted by acceptors are completely unpolarized, so that he is able to derive an equation for the polarization as a function of the concentration both for the low-concentration case and the high-concentration case. In the low-concentration limit, only the interaction between the primary molecule and one other is considered, whereas in the high-concentration limit the excitation energy is thought to diffuse away from the primary molecule. Förster also shows polarization-versus-concentration data for fluorescein in glycerin. The agreement between the data and the low-concentration curve is good up to about one-tenth of the critical concentration. Moreover, the data above the critical concentration are close to the curve derived for the high-concentration case. Finally, he briefly discusses the role of energy transfer in photosynthesis in plants where the chlorophyll concentration is about 0.1 Molar and notes that according to the theory the energy of a photon can migrate over about 10,000 chlorophyll molecules after it is absorbed by the primary molecule. His 1959 review [12] is the 10th Spiers Memorial Lecture and a translation of an earlier paper in German (Th. Förster, Z. Electrochem. 53(1949)93-104). This review is very well written and provides a conceptual understanding of FRET with a minimum of equations. He mentions the classic Bowen papers showing that FRET is different from trivial reabsorption. He compares FRET, reabsorption, donor-acceptor complex formation, and collisional quenching. He mentions three possible situations when his theory is not valid: (1) when thermal relaxation is slow, for example, in gases at low pressures, so that transfer may take place directly from the vibrational level obtained by excitation. In this case the transfer will depend on the excitation wavelength; (2) when dipole-dipole interactions are weak because of forbidden transition in the donor or acceptor; and (3) when the donor-acceptor distances are too small. This last possibility is the Dexter transfer mechanism [16].

His 1949-paper has not been translated into English. It focuses on hetero-FRET, and introduces an extension of his previous theory that predicts the time dependence of the donor fluorescence and the relation between the quantum yield of the donor and the acceptor concentration [13]. He assumes that the rate of molecular rotation is much faster than the rate of transfer, whereas the lateral diffusion is so slow that the donor-acceptor distance does not change during the transfer time. Under these conditions it is shown that the relative donor quantum yield depends only on one variable, the ratio of the acceptor concentration and the critical donor concentration. The predicted relationship between the relative donor quantum yield and this ratio was found to be in good agreement with transfer data for trypaflavine as donor and rhodamine N as acceptor in methanol. Förster uses a very clever way of excluding the effects of reabsorption in his quenching measurements: He uses a large cuvette and a small cuvette with matching concentrations such that for each data point the product of concentration times path-length is the same for both cuvettes. Consequently, the effects of reabsorption are the same in both cuvettes as long as Beer’s law holds. Therefore, the observed differences are due to FRET only.
His 1965-review [14] is not only about FRET. It is a general review of absorption, excitation, and emission. Förster distinguishes three cases. Case A refers to major alterations in absorption spectra, B to less profound changes in absorption spectra, and C to few or no alterations in these spectra. Only case C is relevant for FRET. This is the case of very weak coupling between the excited states of different molecules, where Fermi’s golden rule applies. The excitation in this case can be regarded as temporarily completely localized and excitation transfer can occur. In this review [14] he also presents a modified version of this 1948-theory [11].
His book was published in 1951 [15]. It is a superbly-written overview of fluorescence in organic substances. It describes experimental set-ups used back then. It provides data on FRET and describes the theory in great detail. One of the chapters explains how his equations can be derived from classical electromagnetic theory only, completely without quantum mechanics. This accomplishment is remarkable, because he was expected to spread the word that quantum mechanics is drastically needed in spectroscopy. Who would even consider searching for a classical derivation in his situation? There were rumors that Förster contacted radio engineers. Nobody seems to know why. One can speculate that he understood that FRET is not purely a molecular phenomenon, but that it can be scaled up to an electronic application, similar to what is now known as Near-Field-Communication. Even though FRET can be understood without quantum mechanics, an appreciation of quantum phenomena is still needed as the details of spectra and other relevant concepts in FRET can only be described using the language of quantum mechanics.

When Theodor Förster began his work on energy transfer in the late 1940‘s a significant amount of experimental information on fluorescence and quenching of fluorescence was available [1]. Even a starting point for his theory was available: Franck’s principle. This principle states that if effective energy transfer is to take place from initially excited molecules to quenching molecules, the excited states of the quenchers must be in energy resonance with the primarily excited states [17]. Oppenheimer and Arnold had pointed out that the phenomenon of resonance energy transfer, the basis of FRET, is very similar to internal conversion in radioactivity where an excited nucleus transfers energy without radiation to one of the orbital electrons resulting in ejections of this electron [4, 18, 19]. Using this similarity, they derived an expression of the rate of energy transfer for the case where the acceptors are randomly distributed around a donor. Clegg [4] showed that modifying this expression for the rate of transfer from one dominant frequency to a spectrum of frequencies leads to Förster’s famous equation for the donor-acceptor distance at which the rate of transfer equals that of donor emission, equation 3 below. However, the fact is that Oppenheimer and Arnold did not make these modifications. They did not come up with the idea of incorporating experimentally obtained spectra into their theory. Förster did [10 through 15]. This is what sets Förster apart from his predecessors and contemporaries. They all assumed one dominant frequency and ignored experimental data on spectra. Förster’s most important innovation is to incorporate experimentally obtained parameters such as spectra, quantum yield, and lifetimes into his theory, making it refutable, accessible, and extremely useful.
3 Concepts, Definitions and Equations
The “radio-analogy” is this: Emission of a photon by a molecule is very similar to broadcasting a signal from a radio sender. Absorption of a photon is like picking up a radio wave on your receiver. There are obvious differences in scale and frequencies, but the similarities are clear. FRET is analogous to NEAR-FIELD-COMMUNICATION. This technique actually uses magnetic induction and coils whereas in FRET oscillating electric dipoles interact, and here also, scale and frequencies are different, but the similarities may be helpful for building a conceptual understanding. Resonance plays an important role. Resonance is the phenomenon in which an oscillating system or an external oscillating force drives another system to oscillate with greater amplitude at special frequencies. At such frequencies (called ‘resonant frequencies”) the response is at a maximum, and small periodic driving forces can produce oscillations at large amplitudes. The simplest example is resonance in a swing, which has a natural frequency for going back and forth. Pushing the swing at this resonant frequency yields efficient transfer of energy. In FRET the resonance is electromagnetic. The excited donor contains electrons oscillating around positive centers at frequencies of the order of 10[math]^{15}[/math] Hertz. In Förster’s theory, these oscillating dipoles in the donor set up electromagnetic field and acceptors located in that field interact via resonance.
3.1 Near Field and Far Field



The space around these oscillating dipoles can be considered to consist of 4 parts: the contact zone (Dexter zone), the Near Field, the Intermediate Field and the Far Field (also called the Radiation Field). The contact zone is the space within about 1 nanometer from the oscillating dipole, the Near Field is the space within about 1 to 10 nanometers from it, the intermediate Field is about 10 to 1000 nanometer from it and the Far Field is beyond that. The Electric Field lines generated by an oscillating dipole form loops that are connected to the dipole in the Near Field except on the line through the dipole along the oscillation direction. These electric field lines are straight (see Figure 8). In the Intermediate Field the Electric Field lines are starting to pinch off and the pinching off is complete in the Far Field as illustrated in Figures 7 and 8.
These pinched off loops that are no longer connected to the dipole itself correspond to electromagnetic radiation, that is, photons. FRET happens in the Near Field. Absorption takes place in the Far Field (It is possible to construct a unified description of both processes, see [1] for further reading). Acceptor molecules can be anywhere. In Figure 8 two acceptors are shown (depicted as “A”), one in the Far Field and one in the Near Field. An acceptor in the Far Field is capable of absorbing radiaton emitted by the donor (“D” in Figure 8). Absorption and Emission of electromagnetic radiaton were already very well understood when Förster started his work. The absorption spectrum of the Acceptor must overlap with the emission spectrum of the Donor, otherwise the Acceptor cannot absorb this radiation (this is also resonance). The stronger the overlap the better the acceptor can pick up radiation from the donor.
An acceptor in the Near Field is capable of interacting with the electromagnetic field generated by the donor via FRET. The electric field generated by an oscillating dipole consists of three contributions, one that is proportional to 1/distance, another proportional to 1/distance-squared, and a third proportional to 1/distance-to-the-third-power [5]. The first is dominant in the Far Field and the third is the leading term in the Near Field. Here “distance” means the distance between the oscillating dipole and the point where the field exists. The electromagnetic forces in the Near Field are, therefore, very different from those in the Far Field. However, the frequency-dependence, that is, the spectral aspects of the electromagnetism generated by the excited donor in the Far Field is the same as that in the Near Field. Förster realized that he should incorporate this similarity in his theory of FRET. This idea was probably the secret of his success. The quantity that embodies this idea is R[math]_{0}[/math], the Förster distance, which is that distance at which FRET and donor fluorescence (or, actually deactivation of the donor excited state by means other than FRET) are equally probable. R[math]_{0}[/math] is defined in Equations 3 and 3A. As far as the sizes of the various zones is concerned, Figure 8 gives a rough idea. Figure 9 shows more details: the sizes of the zones depend on wavelength and the refractive index of the medium.
3.2 Quantities in Förster’s Equations
To observe FRET the following conditions must be met:
1) Donor and acceptor must have strong electronic transitions in the UV, visible, or IR,
2) Spectral overlap must exist between donor emission and acceptor absorbance,
3) Donor and acceptor must be close, but not too close,
4) The orientation factor (kappa-squared) should not be too small,
5) The donor emission should have a reasonably high quantum yield,
6) Thermal relaxation must be fast enough so that transfer takes place from the lowest vibrational level of the excited state,
7) dipole-dipole interactions must be strong enough so that forbidden interactions in donor or acceptor are irrelevant.
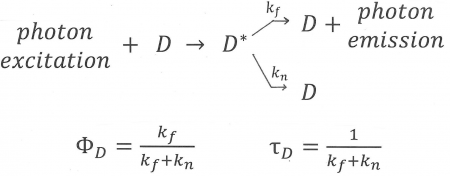
Figure 10. An illustration of the fluorescence process of the donor, D, which enters an excited state, D*, after absorption of a photon. If FRET cannot take place, there are two ways for the donor to lose its excess energy and return to the ground state: by emission (fluorescence) described by rate constant, k[math]_{f}[/math], and by a radiationless transition expressed by rate constant k[math]_{n}[/math]. Equations for the average time the donor remains excited (“lifetime of the donor in the absence of acceptor”) and the “Quantum Yield of donor fluorescence in the absence of acceptor” are also shown.
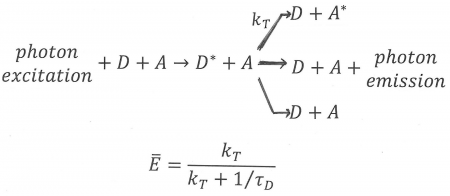
Figure 11. An illustration of FRET in the case of hetero-transfer, described by rate constant kT. Donor, D, and acceptor, A, are both present. First the donor enters an excited state, D*, after absorption of a photon. Next, FRET can take place shown here as a reaction from excited donor + acceptor to de-activated donor + excited acceptor. Note that FRET is always in competition with at least two other processes: donor emission and de-activation of the donor via a radtionless transition, both of which are shown in Figure 11. An equations for the average FRET efficiency is also shown in terms of the rate of transfer and the lifetime of the donor in the absence of acceptor, defined in Figure 11.


The following are the key quantities in Förster’s theory:
- [math]k_{T}[/math]= rate of energy transfer, indicating how often per unit of time transfer can take place; its unit is 1/second or 1/nanosecond or any convenient inverse unit of time,
- [math]\tau_{D}[/math]= lifetime of the donor excited state in the absence of acceptor, the average time the donor is excited when FRET cannot happen, often of the order of a few nanoseconds,
- [math]r_{DA}[/math]= separation distance between donor and acceptor, a center-to-center-distance, often expressed in nanometers,
- [math]R_{0}[/math]= Förster distance = Förster radius (often in nanometers) = that value of the donor-acceptor-separation at which [math]k_{T}=\frac{1}{\tau _{D}}[/math], so that at that particular distance the probability of transfer is equal to the combined probabilities of deactivation of the donor excited state by all other means,
- [math]c_{0}[/math]= critical concentration = that concentration at which transfer is equally probable to other forms of deactivation of the excited donor state,
- [math]\bar{E}[/math] = Efficiency of transfer = the most readily accessible measure of FRET can be obtained by comparing spectroscopic properties in the presence and absence of FRET. It can be determined by at least 5 different ways: by donor quenching, by acceptor sensitization, by a combination of these two, by donor photobleaching, and by acceptor photobleaching [20]. Note there is a bar above the “E”, indicating an average. In ‘Single-Molecule-FRET it is possible to measure efficiency distributions [21]. An efficiency value which is part of a distribution is indicated as [math]E[/math],
- [math]J[/math]= overlap integral, a measure of the level of resonance, defined in detail in equation 6,
- [math]\kappa ^{2}[/math]= orientation factor, which can vary between 0 and 4, describes the interaction of the transition dipoles of donor and acceptor, as explained in Figures 12 and 13 (the possible average values of this are also relevant as explained in 3.4,
- [math]\Phi _{D}[/math]= quantum yield of the donor fluorescence in the absence of acceptor = the ratio of photons emitted by the donor to the number of photons absorbed by the donor,
- [math]n[/math]= the refractive index of the medium in which donor and acceptor are embedded.
3.3 Conditions to Observe FRET
To observe FRET, the following condition must be met:
- The donor and acceptor chromophores have strong electronic transitions in the near ultraviolet to near infrared spectral range,
- There is considerable overlap between the donor emission spectrum and the acceptor absorption spectrum as illustrated in Figure 3,
- The donor and acceptor are relatively close to each other, but not too close; the donor-acceptor separation should be roughly in the 1-10 nanometer range,
- The donor emission moment, the acceptor absorption moment, and their separation vector are in favorable mutual orientation; that is, the orientation factor, kappa-squared, should not be near zero,
- The emission of the donor has a reasonably high quantum yield,
Note that the second condition is also a requirement for transfer by reabsorption. Therefore, it is often necessary in FRET studies to correct for reabsorption effects.
3.4 Equations
Förster’s theory yield the following set of equations:
[math]\bar{E}=\frac{k_{T}}{k_{T}+\frac{1}{\tau _{D}}}=\frac{R_{0}^{6}}{R_{0}^{6}+r_{DA}^{6}}\tag{1}[/math]
[math]k_{T}=\frac{1}{\tau _{D}}\left (\frac{R_{0}}{r_{DA}} \right)^{6}\tag{2}[/math]
[math]R_{0}^{6}=\frac{9(\ln 10)\kappa ^{2}\Phi _{D}J}{128\pi ^{5}n^{4}\tau N _{A}}\tag{3}[/math]
[math]c_{0}\; for\; homo-transfer=\frac{3000}{4\pi N_{A}R_{0}^{3}}\tag{4}[/math]
[math]c_{0}\; for\; hetero-transfer=\frac{3000}{2\pi^{\frac{2}{3}}} N_{A}R_{0}^{3}\tag{5}[/math]
It is a good idea to simplify equation 3 by substituting in the values of the constants creating a “practical form” of this equation. Braslavsky et al have pointed out that there are mistakes in such practical forms all over the literature and recommend including units explicitly [22]. For example, the Förster distance can be written as in equation 3A:
[math]\frac{R_{0}}{nm}=0.02108\times \left [ \frac{\kappa ^{2}\Phi _{D}}{n^{4}}\left ( \frac{J}{M^{-1}cm^{-1}nm^{4}} \right ) \right ]^\frac{1}{6}\tag{3A}[/math]
Here the unit [math]M[/math] is [math]Molar=moles/liter=mol\times dm^{-3}[/math]. An alternative form of equation 2, often used in photosynthesis is:
[math]k_{T}=\left ( \frac{\kappa ^{2}}{r_{DA}^{6}} \right )\left ( \frac{1}{n^{4}} \right )c_{DA}\tag{2A}[/math]
with
[math]c_{DA}=\frac{9(\ln 10)\Phi _{D}J}{128\pi ^{5}\tau _{D}\tau N _{A}}\tag{2B}[/math].
This formulation of the transfer rate establishes a clear separation between geometric properties (kappa-squared/distance-to-6th-power), environmental factors (refractive index) and spectral properties ([math]c_{DA}[/math]). When [math]r_{DA}[/math] is in nanometers and [math]k_{T}[/math] is in inverse picoseconds, [math]c_{DA}[/math] is in nanometer[math]^{6}[/math]/picosecond. Another method of splitting the orientation factor off from the Förster distance is to introduce R-zero-bar:
[math]R_{0}^{6}=\frac{2}{3}\kappa ^{2}\bar{R_{0}^{6}}\tag{3B}[/math]
where [math]R_{0}[/math]-with-bar-on-top is defined as the Förster distance when kappa-squared equals its average value, 2/3. Kappa-squared is explained in Figures 12 and 13. Figure 14 illustates characteristics of equation 1 and 3.
3.5 Orientation Factor and Overlap Integral
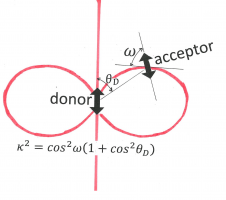
Figure 12. The orientation factor, kappa-squared, depends on the relative orientations of the donor “atomic-antenna” (transition moment, depicted as a double-headed arrow in the center), that of the acceptor “atomic-antenna” (the other double-headed arrow), and that of the line connecting their centers (line connecting the centers of the arrows). The angle [math]\theta _{D}[/math] is between the donor “antenna” and the connection line. The angle [math]\omega[/math] is between the connection line and the electric field generated by the donor at the position of the acceptor. This factor is given given as [math]\kappa ^{2}=\cos ^{2}\omega (1+\cos ^{2}\theta _{D})[/math]. Its value is between 0 and 4. Kappa-squared equals 0 whenever the acceptor “atomic-antenna” is perpendicular to the electric field line (in the plane of the paper or perpendicular to that plane), and it is 4 if both donor and acceptor are parallel to the connection line.
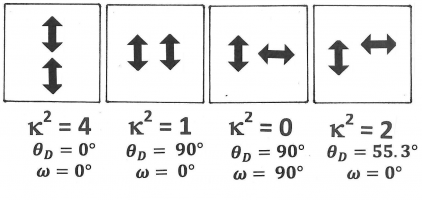
Figure 13. A few specific examples of configurations with different kappa-squared-values. Both donor and acceptor transition moments are indicated as double-headed arrows. It is not necessary to show which of the two is the donor or the acceptor, as kappa-squared is invariant when the donor transition moment and the acceptor transition moment are interchanged.



The overlap integral is illustrated in Figure 15, and depends on wavelength,[math]\lambda[/math], the donor fluorescence spectrum normalized on the wavelength scale, [math]f_{D}(\lambda)[/math] , and [math]\epsilon _{A}(\lambda )[/math] the molar extinction coefficient of the acceptor,
[math]J=\int f_{D}(\lambda )\epsilon _{A}(\lambda )\lambda ^{4}d\lambda\tag{6}[/math]
This integral extends over the region that encompasses the line shapes of the relevant donor emission and acceptor absorption bands. A convenient unit for the overlap integral is OLI = 10[math]^{14}[/math]M[math]^{-1}\lt?math\gtcm\ltmath\gt^{-1}[/math]nm[math]^{4}[/math] = 10[math]^{-14}[/math]M[math]^{-1}[/math]cm[math]^{3}[/math] [1,23]. Wavelength is most often expressed in nanometers, is usually in M[math]^{-1}[/math]cm[math]^{-1}[/math], and has the unit nm-1 and is defined by equation 7:
[math]f_{D}(\lambda )=\frac{F_{D\lambda }(\lambda )}{\int F_{D\lambda }d\lambda } \tag{7}[/math]
where [math]F_{D\lambda }[/math] is the donor fluorescence per unit of wavelength interval and the integral extends over the relevant donor emission band(s). The overlap integral can alternatively be expressed in wavenumber form or in frequency form [1].